Data integrity issues in pharmaceuticals industry: Common observations, challenges and mitigations strategies
Data integrity (DI) reaffirms the pharmaceutical industry’s commitment to manufacture drugs that are safe, effective and fulfill quality standards. At the same time, DI is a crucial tool for regulatory authorities to use in protecting public health. Recent FDA Form-483 observations and warning letters indicate that DI is the main issue the pharmaceutical industry is currently dealing with. Failure to comply with DI requirements may result in a high number of un-validated results, which may cause post-marketing issues and frequent product recalls. To address the underlying causes of DI problems, a comprehensive approach is necessary. The majority of DI issues are caused by
- poor quality culture
- organizational or individual behaviour
- leadership
- processes
- technology
DI should be effectively integrated into the quality management system, and it should apply to both paper and electronic records. Employees should be trained on 21 CFR Part 11. Consistent review and audit are required to ensure that procedures are followed and audit trails are generated.
Electronic system platforms (Data integrity issues in pharmaceuticals)
Electronic systems, in addition to being an efficient solution (system integration, data verification at both input and output, security), offer advantages over traditional paper-based systems in terms of improved compliance with DI regulatory requirements. For example, many electronic system platforms provide enhanced security features and audit trail capabilities.
Electronic system platforms (Data integrity issues in pharmaceuticals)
Finally, management support for data governance is essential for successful implementation of DI. This article reviews commonly observed deviations by FDA pertaining to DI and discusses measures to be undertaken to avoid them.
The data collected during the manufacturing of a pharmaceutical dosage form in a Current Good Manufacturing practice (cGMP) setting must be
accurate and reliable.
cGMP regulations advocate a flexible approach based on risk assessment, depending primarily on process understanding and knowledge management, to avoid and detect data integrity (DI) risks. In this regard, pharmaceutical companies must adopt relevant and effective strategies to fulfil regulatory requirements.
A relevant and effective initiative will consider the facility’s design and operations, as well as advocating system and control monitoring based on the risk to product critical quality attributes, which represent a direct risk to the patient.
Moreover, an effective system design and controls would allow for the easy detection of errors, omissions, and inconsistencies throughout the data life cycle.
The role of management is critical in avoiding and addressing situations that contribute to DI problems. The quality system may fail and stop adhering to cGMP standards in the absence of support from the management system. Management must create a quality culture and make DI a key organizational value, encouraging people to identify and report DI concerns as soon as they arise.
According to 501(a)(2)(B) of the FD&C Act, a drug is deemed adulterated if:
“the methods used in, or the facilities or controls used for, its manufacture, processing, packing, or holding do not conform to or are not operated or administered in conformity with current good manufacturing practice to assure that such drug meets the requirement of the act as to safety and has the identity and strength, and meets the quality and purity characteristics, which it purports or is represented to possess”.
Data suggests that one in 10 medical products in developing countries is either substandard or falsified. Between 2013 and 2017, 62 % of drug shortages in the United States were caused by manufacturing or product quality issues. In this context, the importance of DI cannot be overemphasised.
DI reaffirms the pharmaceutical industry’s commitment to manufacture drugs that are safe, effective and fulfil quality standards.
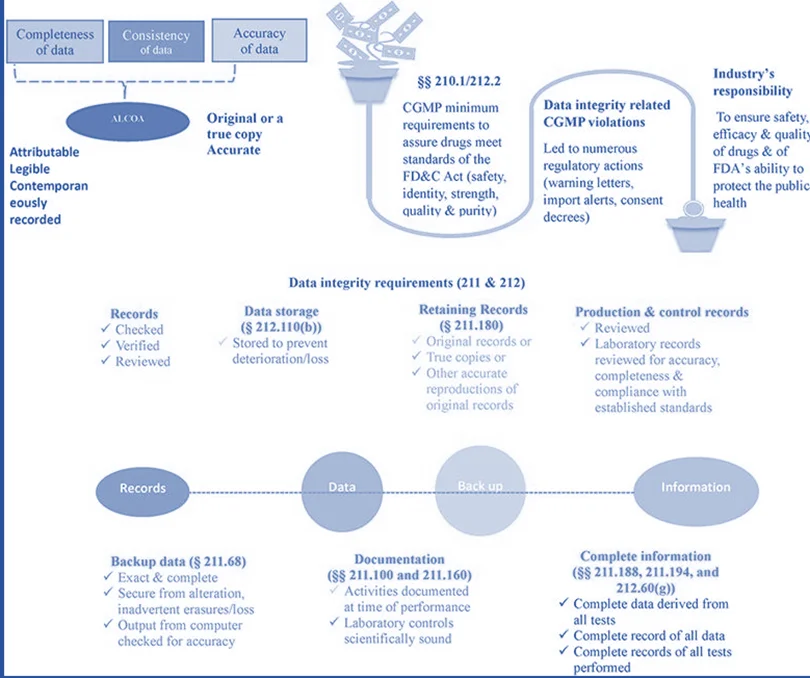
At the same time, DI is a crucial tool for regulatory authorities to use in protecting public health. DI refers to the data that is complete, consistent, and correct, and that is attributable, legible, contemporaneously recorded, original or a true copy, and accurate (ALCOA+). These characteristics (ALOCA+) must be preserved across the course of data’s life cycle, meaning that DI must be preserved during all stages of data including creation, modification, processing, maintenance, archiving, retrieval, transmission, and disposal after the record’s retention period ends.
DI breaches of cGMP have resulted in several regulatory measures, such as:
- The issuance of warning letters, import warnings, and consent decrees.
- The Code of Federal Regulations (CFR) provides the basic cGMP criteria to ensure pharmaceuticals fulfil the standards for safety, identification, strength, quality, and purity.
- In October 2018, the US Food and Drug Administration (FDA) Center for Drug Evaluation and Research (CDER) and the United Kingdom Medicines and Healthcare products Regulatory Agency (MHRA-UK) held a joint Good Clinical Practice (GCP) workshop to review data integrity and clinical data management. It was concluded that the
significance of DI cannot be overstated, and that issues concerning data reliability can have a negative impact on data acceptability submitted in support of marketing authorisation applications. DI violations pose serious safety risks to human volunteers participating in clinical trials and impedes regulatory efforts to protect human subjects during clinical trials. It is important to carefully design and maintain the procedures for keeping the study blind.
- ICH guidance “Guideline for good clinical practice E6(R2)” also recommends assessing DI risks of the clinical trials.
While the FDA guidance clarifies the basic regulatory criteria for DI by providing answers to various questions, the PICS guide outlines the essential components of a successful risk-based DI strategy. Integrating and harmonizing these guidelines would be prudent to promote harmonized DI assessment.
From 2017 till 2022, the FDA has issued more than 160 Warning Letters citing DI deficiencies (FDA, 2022, Warning Letters). In 2022 alone, already 13 Warning Letters have been issued. This article reviews commonly observed deviations by FDA pertaining to DI and discusses measures to be undertaken to avoid them.
The FDA database, primarily Forms 483 and Warning Letters, was searched for DI-related observations. To facilitate discussion, the observations were further classified as data retention, out of specification, metadata, and audit trail, testing into compliance, contemporaneous, aborted runs, original accurate and available, and access to computer systems.
Data integrity mitigation strategies (Data integrity issues in pharmaceuticals)
To ensure data security and reliability, companies should implement data management policies (data governance) and integrate them into the quality system.
Data governance is defined as the overall arrangements that ensure DI.
It can operate through two types of controls:
- Organizational
- technical
Controls such as instructions for record completion, retention of completed records, staff training, authorisation for data generation/approval, data governance system design, constitue organizational data governance.
Technical data governance focuses on the practical, implementation-level aspects of ensuring data quality, security, and availability.
Conclusion of Data integrity issues in pharmaceuticals
Recent FDA Form-483 observations and warning letters indicate that DI is the main issue the Data integrity issues in pharmaceuticals is currently dealing with. Failure to comply with DI requirements may result in a high number of un-validated results, which may cause post-marketing issues and frequent product recalls. Companies that don’t comply with DI requirements receive warning letters, and enforcement actions will have a serious financial impact on them.