


Learn stability-indicating method development, validation, forced degradation studies, ICH Q2(R2) compliance, and pharmaceutical applications.
Definition
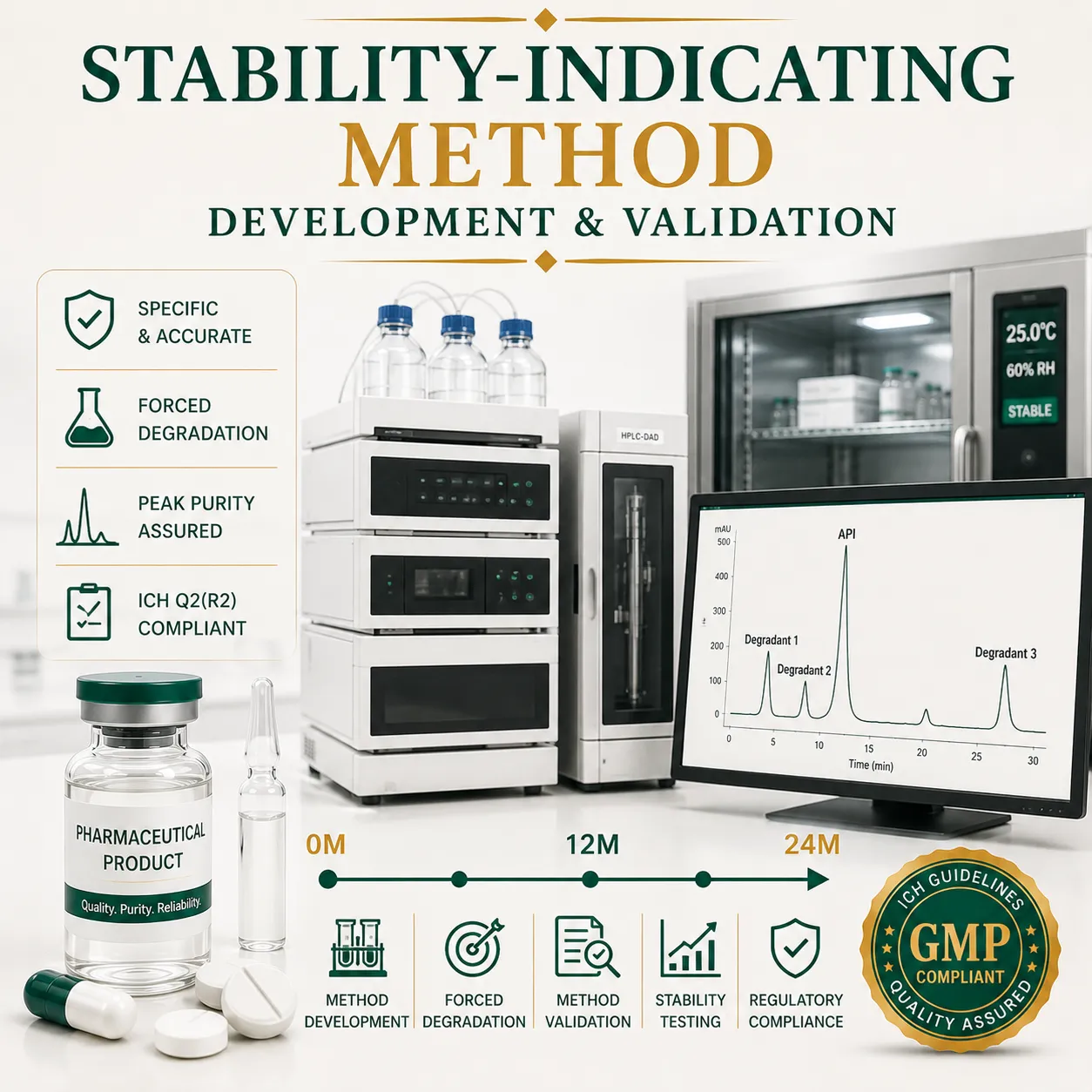
A Stability-Indicating Method (SIM) is a validated analytical procedure that accurately measures an active pharmaceutical ingredient (API) while separating and quantifying degradation products, impurities, and excipients. Stability-indicating methods are essential for forced degradation studies, stability testing, shelf-life determination, and regulatory submissions under ICH Q1A(R2) and ICH Q2(R2) guidelines.
Pharmaceutical products are expected to maintain their identity, strength, quality, purity, and performance throughout their shelf life. To demonstrate this scientifically, manufacturers rely on stability studies supported by robust analytical methods.
However, standard assay methods are often insufficient because they may not distinguish the active pharmaceutical ingredient (API) from degradation products or process-related impurities.
This is where Stability-Indicating Methods (SIMs) become essential.
A stability-indicating method is specifically designed and validated to separate, detect, and quantify the API in the presence of all potential degradation products, impurities, excipients, and formulation matrices.
Regulatory agencies such as the FDA, EMA, MHRA, and WHO expect validated SIMs to support product development, stability programs, quality control testing, and regulatory submissions.
This guide explains the complete lifecycle of stability-indicating methods, including development strategies, validation requirements, practical applications, and GMP considerations.
What Is a Stability-Indicating Method?
A Stability-Indicating Method (SIM) is an analytical procedure capable of accurately measuring changes in a drug substance or drug product over time while ensuring complete separation of the API from all degradation products and impurities.
Primary Objectives of a SIM
- Quantify API potency
- Detect degradation products
- Support stability studies
- Establish shelf life
- Demonstrate product quality
- Meet regulatory requirements
Why Stability-Indicating Methods Matter
| Benefit | Purpose |
|---|---|
| Detect Degradation | Identify product instability |
| Separate Impurities | Ensure accurate assay results |
| Support Stability Studies | Generate reliable data |
| Establish Shelf Life | Support expiration dating |
| Regulatory Compliance | Meet FDA and ICH expectations |
| Product Quality Assurance | Ensure patient safety |
Regulatory Framework for Stability-Indicating Methods
Several international guidelines govern SIM development and validation.
Key Regulatory References
| Guideline | Focus Area |
|---|---|
| ICH Q1A(R2) | Stability testing requirements |
| ICH Q2(R2) | Analytical method validation |
| ICH Q3A/B | Impurity testing |
| USP <1225> | Method validation |
| FDA Analytical Procedures Guidance | Regulatory submissions |
| EMA Analytical Validation Guidance | Method suitability |
Method Development Workflow
Developing a stability-indicating method requires a structured approach to ensure all degradation pathways are evaluated and resolved chromatographically.
Step A: Pre-Formulation Information Gathering
Method development begins with understanding the physicochemical properties of the molecule.
Critical Information to Collect
| Parameter | Importance |
|---|---|
| pKa | Mobile phase selection |
| Log P | Retention behavior |
| Solubility | Sample preparation |
| UV Maxima | Detector wavelength selection |
| Molecular Structure | Degradation prediction |
| Known Impurities | Resolution requirements |
Literature Review
Review:
- Pharmacopeial monographs
- Scientific publications
- Existing analytical methods
- Manufacturing process impurities
- Known degradation pathways
Step B: Forced Degradation Studies (Stress Testing)
Forced degradation is the foundation of every stability-indicating method.
The goal is to intentionally degrade the API and generate representative degradation products.
Target Degradation Level
Regulatory guidance generally recommends:
5–20% degradation
This level provides sufficient degradation products without complete destruction of the molecule.
Common Stress Conditions
| Stress Condition | Typical Treatment |
|---|---|
| Acid Hydrolysis | 0.1–1 M HCl |
| Base Hydrolysis | 0.1–1 M NaOH |
| Oxidation | 3–30% H₂O₂ |
| Thermal Stress | Elevated temperatures |
| Humidity Stress | Controlled RH exposure |
| Photolytic Stress | UV and visible light |
Purpose of Forced Degradation
Forced degradation studies help:
- Identify degradation pathways
- Establish degradation mechanisms
- Demonstrate peak purity
- Verify method specificity
- Support shelf-life studies
Practical Example
An API exposed to 3% hydrogen peroxide showed:
| Component | Result |
|---|---|
| API Remaining | 88% |
| Oxidative Degradant A | 7% |
| Oxidative Degradant B | 5% |
The chromatographic method successfully resolved all components, confirming stability-indicating capability.
Step C: Chromatographic Method Development
After generating degradants, chromatographic conditions must be optimized.
Instrument Selection
The most common platform is:
RP-HPLC with Diode Array Detection (HPLC-DAD)
Advantages:
- High sensitivity
- Peak purity assessment
- Broad applicability
Column Selection
| Column Type | Application |
|---|---|
| C18 | Most APIs |
| C8 | Moderately hydrophobic compounds |
| Phenyl | Aromatic compounds |
| Cyano | Alternative selectivity |
Mobile Phase Optimization
Variables commonly adjusted include:
- Buffer type
- Buffer concentration
- pH
- Organic modifier percentage
- Acetonitrile content
- Methanol content
Gradient vs Isocratic Elution
Mobile Phase Optimization
Variables commonly adjusted include:
- Buffer type
- Buffer concentration
- pH
- Organic modifier percentage
- Acetonitrile content
- Methanol content
Gradient vs Isocratic Elution
Gradient methods are often preferred for stability-indicating assays because they improve separation of multiple degradants.
Stability-Indicating Method Validation (ICH Q2(R2))
Once development is complete, the method must undergo formal validation.
1. Specificity (Most Critical Parameter)
Specificity demonstrates that the API peak is free from interference.
Specificity Requirements
The method must resolve:
- API
- Process impurities
- Degradation products
- Excipients
- Placebo peaks
Acceptance Criteria
- No co-elution
- Peak purity passes
- Adequate resolution achieved
2. Linearity
Linearity confirms detector response is proportional to analyte concentration.
Typical Range
| Test Type | Range |
|---|---|
| Assay | 80–120% |
| Impurities | LOQ–150% |
Acceptance Criteria
Typically:R2≥0.999
3. Accuracy
Accuracy measures closeness to the true value.
Common Approach
Recovery studies using placebo-spiked samples.
Example
| Spike Level | Recovery |
|---|---|
| 80% | 99.2% |
| 100% | 100.1% |
| 120% | 99.6% |
4. Precision
Precision evaluates repeatability and reproducibility.
Precision Types
| Type | Description |
|---|---|
| System Precision | Multiple injections |
| Method Precision | Sample preparation variability |
| Intermediate Precision | Different days, analysts, instruments |
Typical Acceptance Criteria
%RSD≤2.0%
5. Sensitivity (LOD and LOQ)
Definitions
| Parameter | Purpose |
|---|---|
| LOD | Lowest detectable amount |
| LOQ | Lowest quantifiable amount |
Typical Calculation
LOD=S3.3×σ LOQ=S10×σ
Where:
- σ = standard deviation
- S = slope
6. Robustness
Robustness demonstrates reliability during routine use.
Common Deliberate Variations
| Parameter | Typical Change |
|---|---|
| Flow Rate | ±10% |
| Column Temperature | ±5°C |
| Mobile Phase pH | ±0.2 units |
| Organic Phase | ±2% |
Acceptance Criteria
Method performance remains acceptable despite minor changes.
Practical Pharmaceutical Applications
Stability-indicating methods are used throughout the product lifecycle.
Application Areas
Drug Discovery
Identify degradation pathways early in development.
Forced Degradation Programs
Evaluate molecule stability characteristics.
Formal Stability Studies
Support shelf-life determination.
Routine Quality Control
Verify product quality before release.
Regulatory Submissions
Provide evidence for FDA, EMA, and global approvals.
SIM Throughout the Product Lifecycle
| Stage | Application |
|---|---|
| Discovery | Degradation characterization |
| Development | Formulation optimization |
| Stability Testing | Shelf-life assignment |
| QC Release | Batch testing |
| Post-Approval | Ongoing stability monitoring |
Practical Example: Stability-Indicating HPLC Assay
A pharmaceutical company develops a stability-indicating assay for an oral tablet.
Stress Studies Conducted
- Acid degradation
- Base degradation
- Oxidation
- Thermal degradation
- Photolysis
Results
| Condition | Degradation (%) |
|---|---|
| Acid | 12 |
| Base | 8 |
| Oxidation | 15 |
| Thermal | 5 |
| Light | 4 |
Validation Outcome
- Specificity passed
- Linearity R² = 0.9998
- Accuracy 98–102%
- Precision %RSD < 1.0%
The method was approved for commercial stability testing.
GMP and Regulatory Considerations
Regulatory agencies routinely review:
- Forced degradation reports
- Validation protocols
- Raw chromatographic data
- Peak purity evaluations
- Analytical method lifecycle management
- Data integrity controls
Common Regulatory Observations
Validation Outcome
- Specificity passed
- Linearity R² = 0.9998
- Accuracy 98–102%
- Precision %RSD < 1.0%
The method was approved for commercial stability testing.
GMP and Regulatory Considerations
Regulatory agencies routinely review:
- Forced degradation reports
- Validation protocols
- Raw chromatographic data
- Peak purity evaluations
- Analytical method lifecycle management
- Data integrity controls
Common Regulatory Observations
| Deficiency | Regulatory Concern |
|---|---|
| Inadequate stress testing | Method not stability-indicating |
| Poor degradant resolution | Specificity failure |
| Incomplete validation | Data reliability concerns |
| Missing peak purity data | Regulatory questions |
| Weak robustness studies | Method variability risk |
Step-by-Step Guide to Develop a Stability-Indicating Method
Step 1
Gather physicochemical and impurity information.
Step 2
Conduct forced degradation studies.
Step 3
Generate representative degradants.
Step 4
Develop chromatographic separation.
Step 5
Optimize detector conditions.
Step 6
Verify peak purity and specificity.
Step 7
Validate according to ICH Q2(R2).
Step 8
Apply method to stability studies.
Step 9
Maintain method lifecycle monitoring.
Step 10
Support regulatory submissions.
Best Practices Checklist
| Best Practice | Status |
|---|---|
| Conduct forced degradation studies | ✓ |
| Achieve 5–20% degradation | ✓ |
| Resolve all degradants | ✓ |
| Confirm peak purity | ✓ |
| Validate according to ICH Q2(R2) | ✓ |
| Evaluate robustness | ✓ |
| Establish LOD and LOQ | ✓ |
| Maintain GMP documentation | ✓ |
| Review lifecycle performance | ✓ |
| Ensure data integrity compliance | ✓ |
FAQs
1. What is a stability-indicating method?
A stability-indicating method is a validated analytical procedure that accurately measures an API while separating degradation products, impurities, and excipients.
2. Why are stability-indicating methods important?
They ensure accurate stability testing, support shelf-life determination, and meet regulatory requirements.
3. What is the purpose of forced degradation studies?
Forced degradation generates degradation products to demonstrate method specificity and stability-indicating capability.
4. Which analytical technique is most commonly used for SIMs?
RP-HPLC with diode array detection (HPLC-DAD) is the most commonly used technique.
5. What degradation level is targeted during stress testing?
Typically 5–20% degradation is targeted.
6. What guideline governs SIM validation?
ICH Q2(R2) is the primary guideline for analytical method validation.
7. What is specificity in SIM validation?
Specificity is the ability to separate the API from impurities, degradants, and excipients.
8. What are LOD and LOQ?
LOD is the lowest detectable concentration, while LOQ is the lowest quantifiable concentration.
9. How is robustness evaluated?
By introducing deliberate variations in analytical parameters and assessing method performance.
10. Where are stability-indicating methods used?
They are used in drug development, stability studies, batch release testing, formulation screening, and regulatory submissions.